The Dravet syndrome Spectrum

Introduction
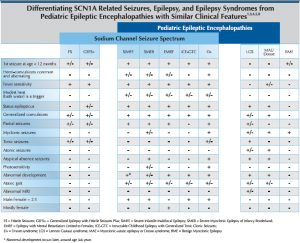
Sodium channel related seizure disorders encompass a spectrum that ranges from simple febrile seizures (FS) and genetic epilepsy with febrile seizures plus (GEFS+) at the mild end to Dravet syndrome (Ds) at the severe end. There are currently 359 SCN1A disease-causing mutations reported in the literature. Recent discovery of the association of SCN9A3 and PCDH194 mutations with pediatric epileptic encephalopathies having similar presentation and prognosis to Dravet syndrome have expanded the realm of the genetic basis and clinical presentation of the spectrum. Consistent genotype/phenotype correlations are not well established and further research is necessary to understand the full spectrum of this genetic epileptic encephalopathy. Truncating and de novo mutations appear to be associated with a more severe disease course. While most SCN1A mutations arise de novo, inherited cases and parental mosiacism have been reported.
![]()
Epidemiology
Ds is a rare disorder with an incidence estimated between 1:20,000 and 1:40,000, representing 3-8%of patients having their first seizure before age one. While clinical diagnosis of Dravet syndrome is determined based on age of onset; seizure presentation; triggers including moderate heat, fever, illness and photosensitivity; genetic confirmation of mutation at any age may guide pharmacologic treatment and will allow evaluation of genotype/phenotype correlations over time.
Differential Diagnosis
Differentiation of Dravet syndrome from febrile seizures or febrile status epilepticus early in the course allows focused therapeutic intervention and leads to improved outcome. Genetic Epilepsy with Febrile Seizures plus (GEFS+) may be caused by an inherited SCN1A missense mutation that most often does not result in an epileptic encephalopathy. Because parental mosaicism is reported and inherited mutations may yield different clinical courses. Children diagnosed with SCN1A mutations should be followed by a developmental psychologist to ensure appropriate developmental milestones are achieved.
Suspicion of Ds is warranted at the second prolonged seizure which is often associated with vaccines, fever or illness in a child with seizure onset before the first birthday. If an infant presents with myoclonic jerks after the first prolonged seizure that occurs before the first birthday, Ds should be considered early in the differential diagnosis. Ruling out focal epilepsy due to lesion and familial diagnosis including familial neonatal seizures, benign familial infantile seizures, benign childhood epilepsy with centrotemporal spikes, child occipital epilepsy and progressive myoclonic epilepsies due to metabolic disorders is worthwhile.
More obvious causes of seizure activity may be warranted before considering a genetic epilepsy diagnosis including: trauma, hypoxia, meningitis, hemorrhage, toxins, endocrinopathy, and B-6 related seizures. Ds can be differentiated from other non-progressive pediatric epileptic encephalopathies with myoclonic seizures, such as Lennox-Gastaut Syndrome (LGS) and Myoclonic Astatic Epilepsy (MAE), by clinical presentation and seizure semiology. While uncommon, LGS and MAE diagnoses may overlap with SCN1A mutations. Overlap with cryptogenic generalized or cryptogenic focal epilepsy and SCN1A mutation may be more common.
Genetic confirmation of a clinical diagnosis of Dravet syndrome is useful for Pharmacotherapy guidance. Avoiding drugs that exacerbate seizures in sodium channel related epilepsies may improve the outcome regardless of age at the time of diagnosis.
Drugs to Avoid (click for details)
Sodium channel blockers including phenytoin, fosphenytoin, carbamazepine, oxcarbazipine, lamotrigine, lascomide and rufinamide are not recommended for acute or chronic management of Ds. These drugs may worsen the seizures cased by sodium channel mutations producing loss of channel function. The correlation between type of mutation and loss or gain of function is not straightforward, therefore avoiding these drugs is prudent. Selective GABA reuptake inhibitor/GABA-T inhibitors including vigabatrin and tiagabine often exacerbate myoclonic seizures and may worsen overall seizure control.
Drugs that are Efficacious in Ds
Ds is refractory and treatment resistant. For acceptable seizure control and to minimize episodes of status epilepticus, most children will require multiple agents – especially in the first five years of life. Stiripentol + clobazam + valproic acid is the most well studied and efficacious regimen for children with Ds. Topirimate, valproic acid, clonazepam, clobazam, levetriacetam, potassium bromide, zonisamide and the ketogenic diet in various combinations have also shown efficacy in Ds.
Stiripentol (Diacomit-Biocodex, France) is approved by teh EMEA for use in conjunction with Clobazam and valproic acid as an adjunctive therapy of refractory generalized tonic-clonic seizures in patients wtih Ds whose seizures are not adequately controlled with Clobazam and valporic acid. Stiripentol works indirectly as a cytochrome P450 coenzyme inhibitor, linearly increasing the area under the curve of drugs with major metabolism through this pathway. It also has a weak GABA-ergic activity. The stiripentol, clobazam and valporic acid combination for treatment of Ds is supported by well controlled clinical trials. Stiripentol is not not indicated as monotherapy. Caution should be exercised when adding stiripentol to clobazam and valproic acid due to significant increases in active drug and metabolites which may lead to toxicity. In the event of clinical signs of side effects or over dosage of clobazam (i.e., drowsiness, hypotonia, and irritability in young children), this daily dose was reduced by 25% every week. Approximately two to three fold increases in clobazam and five fold increases in non-clobazam plasma levels respectivelyhave been reported with co-administration of stirpentol in children with Ds.
The potential for metabolic interaction between stiripentol and valporic acid is considered modest and thus, no modification of valporate dosage should be needed when stiripentol is added, except for clinical safety resons. In the pivotal studies in the event of gastrointestinal adverse reactions such as loss of appetite, loss of weight, the daily dose of valporic acid was reduced by around 30% every week.
Manufacturer recommended doses begin with stiripentol 10mg/kg/day and up to 50mg/kg/day divided two to three times a day with dose adjustments every three days. Lower doses and trough levels of valproic acid are required when stiripentol is utilized to maximise tolerance.
Trials are lacking for use of the stiripentol combination with the ketogenic diet.
Significant drug interactions with all drugs extensively metabolized by the CYP450 system may occur. Drugs to avoid with stiripentol include rye, ergot, alkaloids, cisapride, halofantrine, pimozide, quinidine, bepridil, immunosuppresants, and statins. Precaution should be exercised when using stiripentol with benzodianzepines, theophylline, caffine, chlorpromazine, and other anti-epileptic drugs. Avoidance of macrolide antibiotics is prudent due to their metabolism by the P450 system. Liver function and blood counts should be assessed every 6 months. For more information about drug interactions and dosing please contact Biocodex at or a pharmacist at ICE Alliance by emailing us at [email protected] Clobazam (Frisium-Lundbeck, Chicago) is an atypical benzodiazepine with activity at the 1,5 benzodiazepine receptor. Traditional benzodiazepines bind to the 1,4 benzodiazepine receptor. Clobazam is less sedating and less likely to blunt cognition compared to traditional benzodiazepines.
Manufacturer recommended doses for clobazam are 0.5-1 mg/kg/day once a day or in divided doses in children less than two years old. 16 clinical studies utilizing the combination of stiripentol, clobazam and valproric acid do not support doses of clobazam beyond 0.5 mg/kg/day.
Buccall midazolam maleate-(Epistatus Special Products, London) is available as an unlicensed product in the UK. The dose of buccally or sasally administered midazolam for the treatment of an acute seizure event is 0.3-0.5 mg/kg/dose.
Stiripentol, clobazam and buccal midazolam maleate are not FDA approved and have limited availability outside of Europe. Country specific drug importation regulatory and compassionate use policies may allow importation of these drugs for the treatment of Ds or refractory status epilepticus. Stiripentol can be obtained from Biocodex www.Biocodex.com. Caligor Pharmacy in NY,USA is a drug importer and exporter. Contact Caligor Pharmacy at 212.369.6000 or FAD: 212.879.8415. For more information regarding importing these drugs or insurance, National Formulary, or Medicaid coverage, please contact us at [email protected].
Facial Features of Ds
Dysmorphic features of patients wtih Ds are not reported. Patients with Ds have similar symmetric facial features including large, wide-set eyes; a flat nasal bridge; full lips; low facial muscle tone; and an oval face with a small chin. This “cherubic look”continues into adulthood. Multi-national dysmorphology studies are ongoing.
Co-Morbid Conditions of Dravet syndrome
• Expressive and receptive language impairment
• Pronation of feet that often goes unnoticed and leads to painful orthopedic conditions
• Ataxia and gait abnormalities
• Low muscle tone
• Chronic infection and low humoral immunity
• Growth and nutrition abnormalities including malnutrition and kyphosis
• Dysautonomia
• Autistic spectrum, attention deficit, and other behavioral disorders including psychosis
• Sleep disorders
Prognosis of Dravet syndrome
Outcome of the sodium channel related epileptic encephalopathies hard to predict and a better prognosis is expected when seizures do not become status epilepticus and seizure control is optimized. In Ds, ICE-GTC and SMEB the types and duration of seizures and extent of intellectual impairment , behavioral disturbance, and neurologic impairment manifest by age four years. Intellectual, behavioral and neurologic disturbances may appear later (between 3 and 6 years of age) in SIMFE. EMRF, which is associated with a PCDH19 mutation in females, follows the same prognosis as Ds; however, myoclonic and atypical absence seizures are usually not present. The number of seizures and perhaps more importantly the number of episodes of status epilepticus as an infant and young child is a risk factor for the degree of developmental compromise, behavioral disturbance, and ultimate ability to gain seizure freedom in adulthood. The mortality rate is 16-20% by age 20 and is related to prolonged convulsive seizures, accident and sudden unexplained death. These statistics are based on epidemiologic studies prior to the development of more recent medication regimens such as stiripentol combined with valproic acid and clobazam or topirimate, which provide better seizure control and fewer episodes of status epilepticus in this population and therefore could improve the morbidity and mortality of Dravet syndrome.
Sudden Unexpected Death in Epilepsy
What is sudden unexpected death in epilepsy (SUDEP)?
The definition of SUDEP is “sudden, unexpected, witnessed or unwitnessed, non-traumatic and non-drowning death in epilepsy, with or without evidence for a seizure and excluding documented status epilepticus, in which postmortem examination does not show toxicological or anatomical cause for death.” The death could not be directly caused by seizure or status epilepticus. Also, autopsy does not show any medical cause of death.
Why does SUDEP occur?
It is suspected that SUDEP has to do with involvement of the heart and respiration.
Those who have witnessed SUDEP explain that the patient has breathing difficulties.
How often does SUDEP occur?
Epilepsy affects 50 million people worldwide and SUDEP is the cause of a mean of 17% of deaths people with epilepsy. The reported incidence of SUDEP is quite variable, largely because of differences in patient populations, study design, and the criteria for defining SUDEP. Studies evaluating the incidence of SUDEP among all epilepsy patients within a community report lower incidences (0.13–2.7/1,000 person-years) than those among more selective populations, such as patients being treated at tertiary referral centers (1.2–5.9/1,000 person-years), those undergoing drug or device trials (3.5–6.0/1,000 person-years), and those being evaluated for epilepsy surgery (as high as 9.3/1,000 person-years). Such results imply that patients with fewer seizures carry a lower risk of SUDEP.
Based on limited study reports, there appears to be a lower risk of SUDEP among children. Four studies documented a total of 43 cases of SUDEP in children; the authors estimated the risk of SUDEP in children to range from 0.11 to 0.43 per 1,000 person-years.
The risk factors of SUDEP
There are many suspected risk factors to be involved with SUDEP. There are certain risk factors that have more evidence.
- The risk factor with the strongest link to SUDEP is the seizure frequency.
- It is suspected that as the annual occurrence of seizures increases so doses the risk of SUDEP.
- Generalized tonic-clonic seizures place patients at a higher risk for SUDEP.
- Younger adults and those with cognitive dysfunction may be the most common groups which are affected by SUDEP; however, studies are conflicting on these matters and cause of mortality as SUDEP may be reported more often in young adults compared to older adults.
- Lack of anti-epileptic therapy is a risk factor for SUDEP as is non-compliance.
- Polytherapy with anti-epileptics is a risk factor for SUDEP as drug resistant epilepsy is implied.
- Some studies have associated that the earlier the age of onset of epilepsy is as risk factor for SUDEP.
Prevention of SUDEP
- Maximizing seizure control carries the strongest evidence to reduce risk for SUDEP.
- Night supervision is a key factor in preventing SUDEP. It is important because it is theorized that supervision of the patient can help to prevent seizure associated respiratory complications by being able have to the capability to position the patient. Supervision implies regular checks during throughout the night, use of listening devices, or sharing a bedroom.
- Removal of pillows and loose clothing or blankets during sleep, while not proven, may help prevent SUDEP.
- Omega – 3 fatty acid consumption or supplementation may decrease risk of sudden cardiac death.
References: Curr Neurol Neurosci Rep (2010) 10:319–326
Arq Neuropsiquiatr. 2009 Sep;67(3B):927-9.
Epilepsy & Behavior Volume 14, Issue 1, January 2009, Pages 27-31
Summary
Appropriate diagnosis of sodium channel related encephalopathies and management with appropriate medications and therapies can improve the outcome. Stiripentol + clobazam + valproic acid has the best supporting clinical evidence for the treatment of Ds. Topirimate, zonismide, levetiracetam, potassium bromide, clobazam, clonazepam, valproic acid and the ketogenic diet in various combinations may also provide acceptable seizure control. Phenytoin, fosphenytoin, carbamazepine, oxcarbazapine, lamotrigine, lacosamide, rufinamide, vigbatrin, and tiagabine should be avoided. Treatment in the infant and toddler years will require multiple drugs. Genetic diagnosis of any patient with a seizure onset before age one and presentation consistent with Ds may lead to improved pharmacologic management and qulality of life.
References
- Dravet C, Bureau M, Oguni H. et al, Severe myoclonic epilepsy in infancy: In:Rober J, BureauM, Dravet C, et al (eds) Epileptic Syndromes in Infancy, Childhood and Adolescense, 3 ed. John Libbey, Eastligh, pp 81-103.2002
- Lossin. A catalog of SCN1A variants. Brain Dev 2008:doi; 16:1016jbraindev.2008.07.011
- ILAE
- MillerI, Sotero de Menezes M. SCN1A Seizure Disorders. Gene Reviews: NCBI Bookshelf 2007; www.ncbi.nlm.nih.gov/bookshelf.
- Harkin La, McMahon JM. The spectrum of SCN1A related infantile epileptic encephalopathies. Brain 2007 Mar; 130 (pt 3): 843-52
- Nabbout R Gennaro E et al. Spectrum of SCN1A mutations in sever myoclonic epilepsy of infancy. Neurology. 2003; 60: 1961-7
- KIMURA K, SUGAWARA T, A missense mutation in SCN1A in brothers with sever myoclonic epilepsy in infancy (SMEI) inherited from a father with febrile seizures. Brain &Dev (2005);27: 424-430
- Hurst D.Epidemiology of Severe Myoclonic Epilepsy of infancy. Epilepsia 2001;42:796-803
- Gardiner M, Molecular Genetics of Infantile Nervous System Channelopathies. Early Human Development 2006; doi:10.1016jearlhdev.200609.013
- Scheffer I, Berkovic S, Generalized Epilepsy with Febrile Seizures Plus: A genetic disorder with heterogeneous clinical phenotypes. Brain 1997; 120:479-490
- Scheffer I, et al., Neonatal epilepsy syndromes and GEFS+. Epilepsia 2005:46(10):41-7
- Iwasaki N, Nakayama J. Molecular Genetics of Febrile Seizures. Epilepsia 2002; 43:32-35
- Scheffer I, Berkovic S, Generalized Epilepsies with Febrile Seizures Plus. In:Engle J, Pedley T. Epilepsy: A Comprehensive Textbook. Lippincott Williams & Wilkins 2007; 2553-2662
- GrossoS et al., Efficacy and safety of topiramate in refactory epilepsy of childhood; long-term follow up study. Jour of Child Neur 2005; 20(11):893-97
- Chiron C, et al., Lancet 200; 356:1638-42. 11.
- Yakouh M, et al., Early diagnois of sever myoclonic epilepsy in infancy. Brain Dev 1992:14;299-303.
- Pellock JM. Tiagabine (Gabitril) Experience in Children. Epiepsia 2001;42:49-51.
- Laux, L. Personal communication
- Dravet C. Personal communication
- Caraballo R, Cersosimor R. et al., Ketogenic Diet in patients with Dravt syndrome. Epilepsia 2005; 46:1539-44
- Korff C, Laux L. Dravet syndrome: a retrospective study of 16 patients. Jour Child Neur 2007; 22:185-194